【文章内容】
我们设计了一种以四氢吡喃(THP)为溶剂的弱溶剂化性醚电解质,以实现石墨负极中锂离子(Li+)的可逆快速插层。传统的醚基电解质很容易在石墨层中发生插层,而以四氢吡喃为溶剂的电解质则不同,它具有快速去溶剂化能力,能与石墨负极很好地匹配。此外,Li+ 和 THP 之间的微弱互联允许更多的阴离子进入 Li+ 的溶剂化鞘,从而形成富含无机物的界面,从而抑制了副反应。因此,使用 THP 电解质的LFP/石墨软包电池(3 Ah)在 2 C 充电条件下循环 500 次后,容量保持率为 80.3%,远高于酯电解质系统(200 次循环后为 7.6%)。在 4 C 充电条件下,放电容量从酯类的 2.29 Ah 提高到 THP 的 2.96 Ah。此外,电池还能在较宽的工作温度范围内(-20 至 60 ℃)正常工作。研究人员的电解质设计让人们对快速充电和宽温条件下的锂离子电池有了一定的了解。该成果以题为“A Weakly Solvating Ether Electrolyte Enables Fast-Charging and Wide-Temperature Lithium-Ion Pouch Cells”发表在国际知名期刊ACS Nano上,第一作者为博士生廖亚祺和林文杰。
【研究背景】
石墨基锂离子电池在电动汽车市场取得了巨大成功。然而,基于碳酸酯的传统电解质具有缓慢的去溶剂化动力学过程,在快速充电和低温下它们的性能会恶化。本论文中,设计了四氢吡喃(THP)为溶剂的弱溶剂化醚电解质,锂离子能够在石墨负极中实现可逆且快速的锂离子(Li+)嵌入。与传统的醚基电解质容易共嵌入石墨层不同,THP基电解质表现出快速的去溶剂化能力,并且可以与石墨负极良好匹配。此外,Li+和THP之间的弱相互连接使得更多阴离子进入Li+的溶剂化壳,诱导富含无机物的界面,从而抑制副反应。
【研究亮点】
亮点一:
在这项工作中,研究人员本文设计了一种以四氢吡喃(THP)为溶剂的弱溶剂化醚电解质。与其他环醚相比,THP 的键变形程度更小,因此可以在不发生开环聚合的情况下保持结构稳定,以实现锂离子在石墨负极中的可逆和快速嵌入,优化锂离子电池快充和低温性能
亮点二:
Li+与THP之间的弱溶剂化作用使得更多的阴离子进入Li+的溶剂化壳层,形成富无机界面,从而抑制副反应。
亮点三:
组装Ah级别软包验证电性能,使用基于 THP 的电解质的 Ah 级 LFP/Gr 软包电池具有高倍率性能(4 C 充电时为 2.96 Ah)、长周期稳定性(1000 个周期后容量保持率为 83.1%)和宽工作温度范围(-20 至 60 ℃)。
【图文导读】
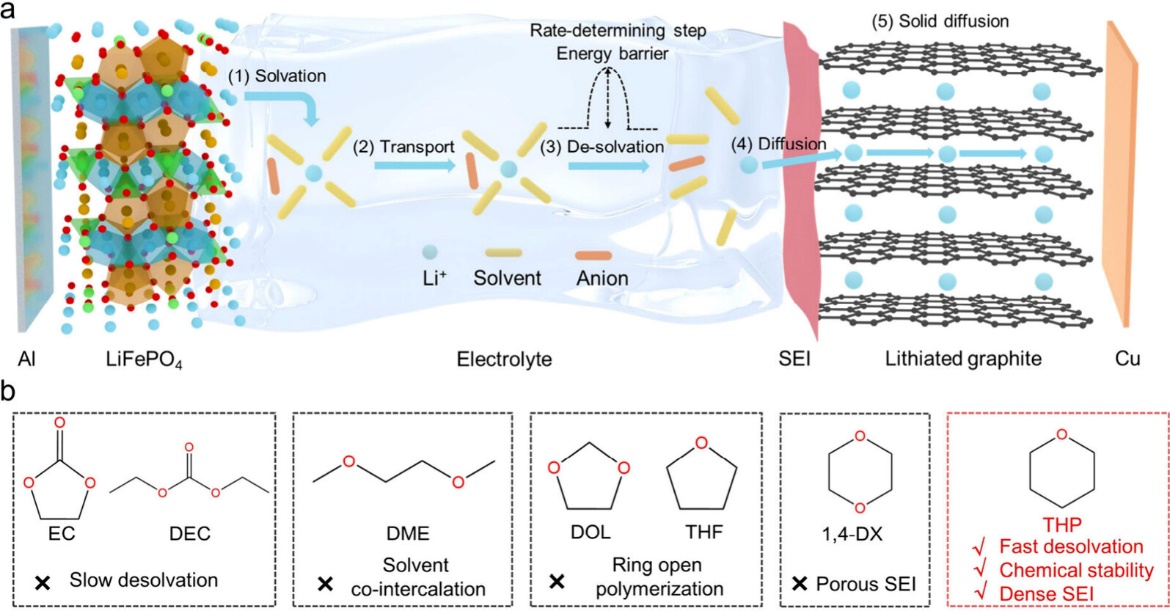
图1. (a) 充电过程中 Li+ 迁移路线示意图。(b) EC/DEC、DME、DOL、THF、1,4-DX 和 THP 溶剂的比较。
如图1a所示,Li+迁移过程在充电过程中包含以下几个步骤:
(1)Li+从正极脱出,自发地与溶剂和阴离子形成溶剂化单元。
(2)Li+-solvent-anion单元在电解质中运输。
(3) Li+-solvent-anion单元在SEI附近脱溶。
(4) Li+在SEI内部扩散。
(5)Li+向石墨层扩散,形成经典的石墨插层产物(LixC6)。
步骤1是放热反应步骤2和5受到电极和电解质固有性质的影响。界面之间的电荷传递过程3和4遇到了显著的动力学障壁。人们普遍认为,脱溶过程比其他步骤具有更高的动力学能垒。与酯类溶剂相比,醚类溶剂由于溶剂化能力弱、脱溶动力学快,更受LIBs快速充电的欢迎(图1b)。传统的线性醚分子(如1,2-二甲氧基乙烷、DME)共插在石墨层中;常用的环醚DOL和THF容易发生开环聚合反应。它们不适合作为高动力学电池电解质溶剂的候选者。THP和1,4-二恶烷(1,4-DX)等六元环醚具有良好的结构稳定性。
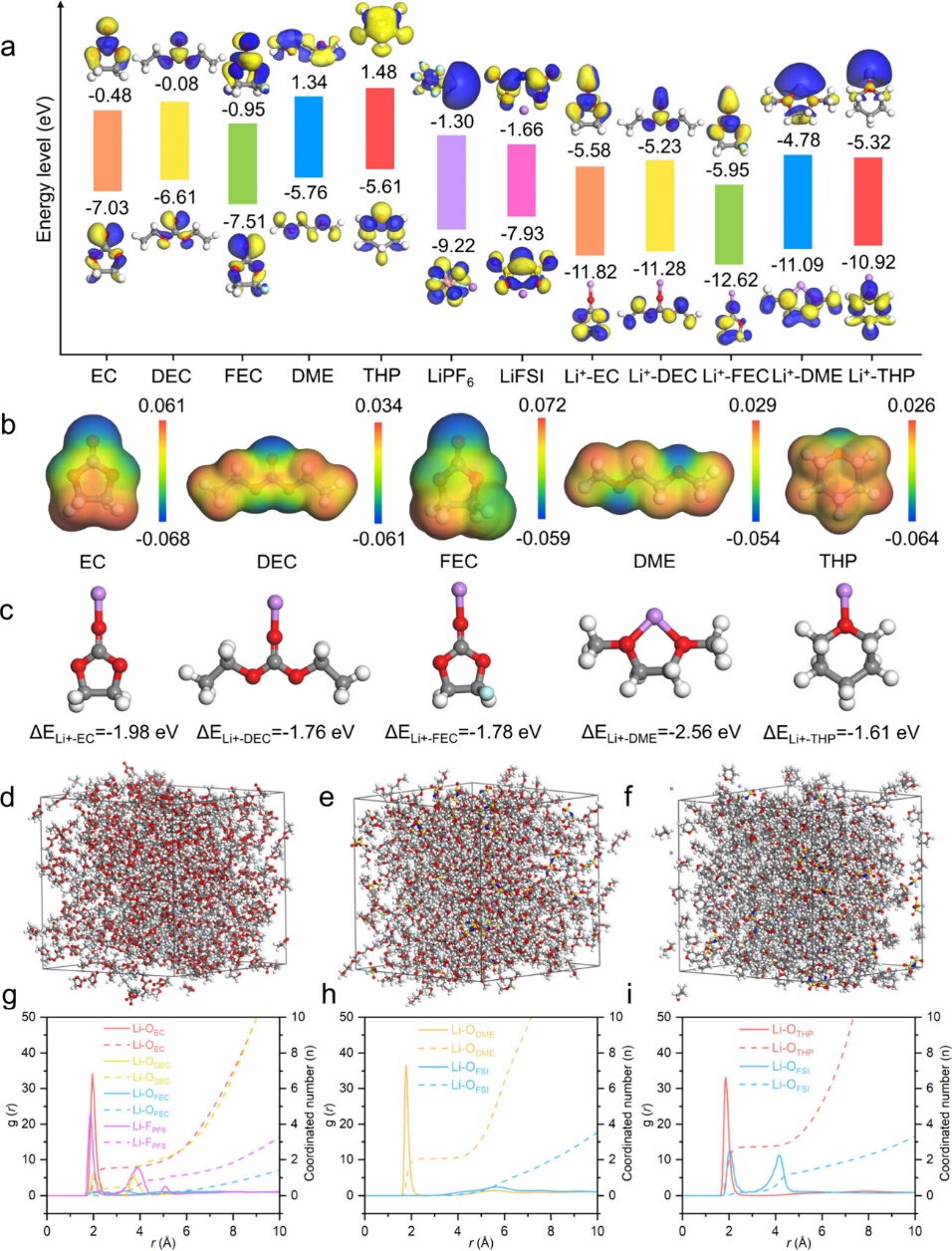
图2. (a) 溶剂、盐和 Li+ 团簇的能级。(b) EC、DEC、FEC、DME 和 THP 分子的 ESP。(c) Li+ -EC、Li+ -DEC、Li+ -FEC、Li+ -DME 和 Li+ -THP 分体相电解质的分子动力学模拟和半径分布函数:(d,g)EC/DEC 中的 LiPF6;(e,h)DME 中的 LiFSI;(f,i)THP 中的 LiFSI。子的结合能。
图2a比较了溶剂、盐和Li+-solvent单元的最高已占据轨道(HOMO)/最低未占据轨道(LUMO)值。EC、DEC、FEC、DME和THP的HOMO/LUMO水平分别为−7.03/−0.48、−6.61/−0.08、−7.51/−0.95、−5.76/1.34和−5.61/1.48 eV。显然,THP在溶剂中表现出最高的LUMO水平,这意味着在负极表面的抗还原能力最好。对于盐,LiFSI的LUMO水平为- 1.66 eV,低于LiPF6 (-1.30 eV),这证实了LiFSI更容易被还原。与Li+配位后,Li+-EC、Li+-DEC、Li+-FEC、Li+-DME和Li+-THP的HOMO/LUMO能级分别降至−11.82/−5.58、−11.28/−5.23、−12.62/−5.95、−11.09/−4.78和−10.92/−5.32 eV,表明配位溶剂比自由溶剂更容易被还原。通过计算醚类化合物的静电势(ESP)来分析其溶剂化能力。如图2b所示,THP的ESP范围较碳酸酯窄,为−0.064/0.026 eV。Li+-EC、Li+-DEC、Li+-FEC、Li+-DME和Li+-THP的结合能分别为−1.98、−1.76、−1.78、−2.56和−1.61 eV(图2c)。电荷密度差结果显示THP和Li+之间的电子转移较少。以上计算验证了THP是一种弱溶剂化溶剂。
通过分子动力学模拟研究了Li+的溶剂化结构。在EC/DEC电解质中,Li+−OEC峰出现在1.95 Å处,表明EC占据了内溶剂化壳层(图2g)。Li+和PF6−被有机溶剂分离,生成经典的Li+溶剂化结构:溶剂分子占据第一溶剂化壳层。在DME电解质中,Li+-ODME峰出现在1.79 Å处,Li+-OFSI峰不存在(图2h),说明DME在第一溶剂化层中占主导地位。与此形成鲜明对比的是,在THP电解质的2.03 Å处观察到Li+-OFSI峰(图2i),表明许多FSI−参与第一溶剂化壳。虽然DME和THP电解质都采用LiFSI作为单盐,但由于THP的溶剂化能力弱于DME,其Li+溶剂化结构不同。
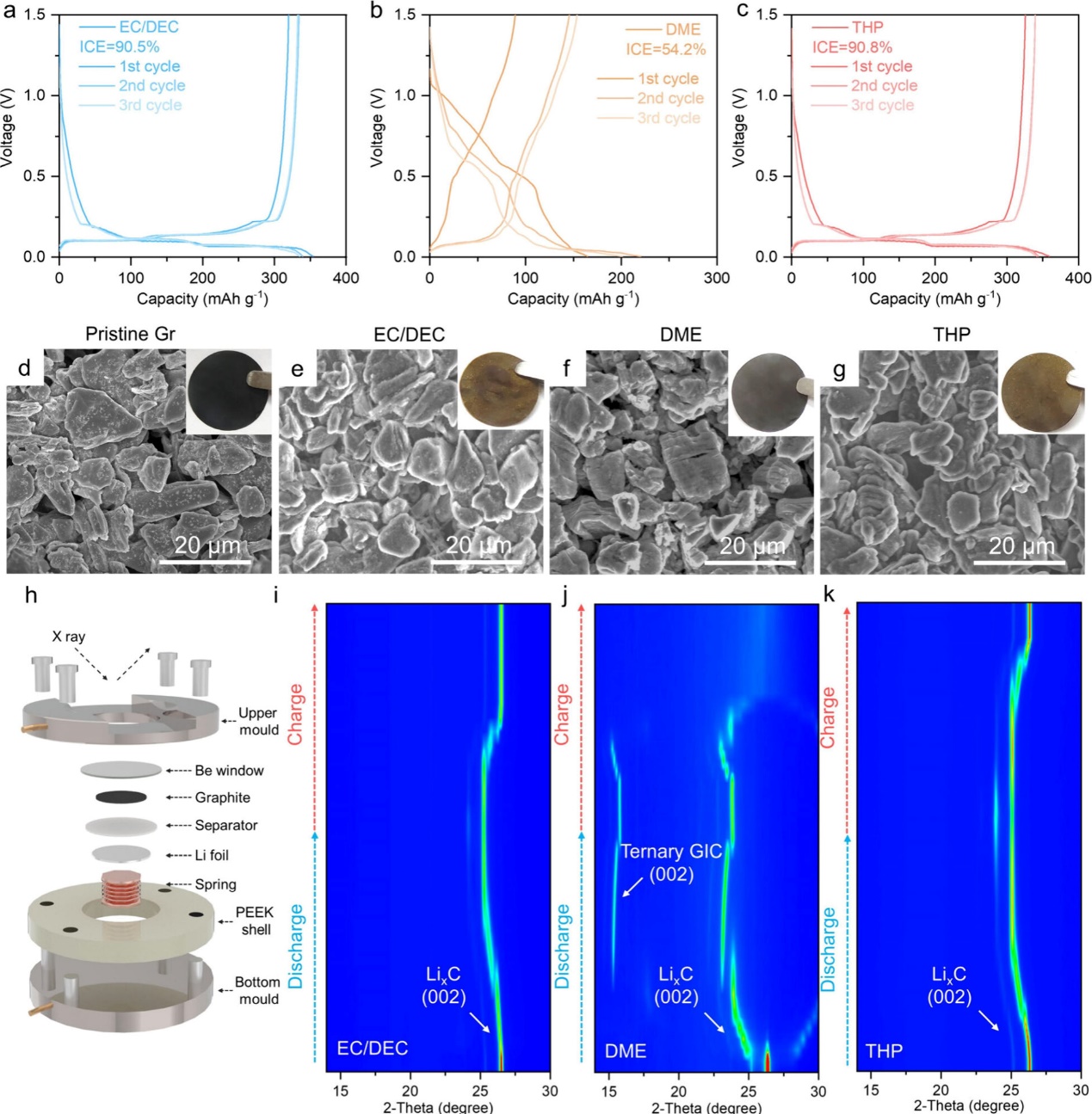
图3. (a) EC/DEC、(b) DME 和 (c) THP Li/锗半电池的放电/充电曲线。(d)原始和在(e)EC/DEC、(f)DME 和(g)THP 中循环的 Gr 电极的 SEM 图像。插入的数字图像为 Gr 电极。(h) (i) EC/DEC、(j) DME 和 (k) THP 电解质中模拟电池的原位 XRD 测试示意图。
组装不同电解质的Li/Gr半电池,研究Li+插层化学。含有EC/DEC、DME和THP电解质的Li/Gr电池的ICEs分别为90.5%、54.2%和90.8%(图3a−c)。除了库仑效率最低外,使用DME电解质的电池在前三个循环中也表现出严重的容量衰减。使用EC/DEC和THP电解质的电池具有相似的放电容量。图3d-g比较了新鲜和循环后的Gr电极的形貌。Gr电极在循环前呈现光滑的层状结构和黑色(图3d)。在DME电解质中循环后,Gr电极显示出结构塌陷,层空间扩大,片状脱落,表明溶剂共插层过程(图3f)。Gr电极仍为黑色,表明形成了石墨插层化合物。相比之下,在EC/DEC和THP电解质中循环的Gr电极保持其层状结构而没有结构崩溃(图3e,g)。电极变成金黄色,对应于LiC6的颜色。为了深入研究Li+插层机理,对Li/Gr半电池进行了原位X射线衍射(XRD)测试。图3h显示了原位XRD组件电池的结构。组件电池的电压曲线与普通扣式电池相似。放电前,在26.5°处有一个视峰,属于Gr的(002)间距,(002)面间距计算为3.36 Å。放电初期,由于Li嵌层的晶格膨胀,(002)峰逐渐向较低的衍射角移动。在DME电解质中,在15.1°处出现一个峰值,与石墨插层化合物相对应(图3j)。结果证实了DME电解质中存在溶剂共插层现象。相比之下,EC/DEC和THP电解质在15.1°处没有观察到峰,这意味着只有Li+(脱)插层而没有溶剂共插层(图3i,k)。
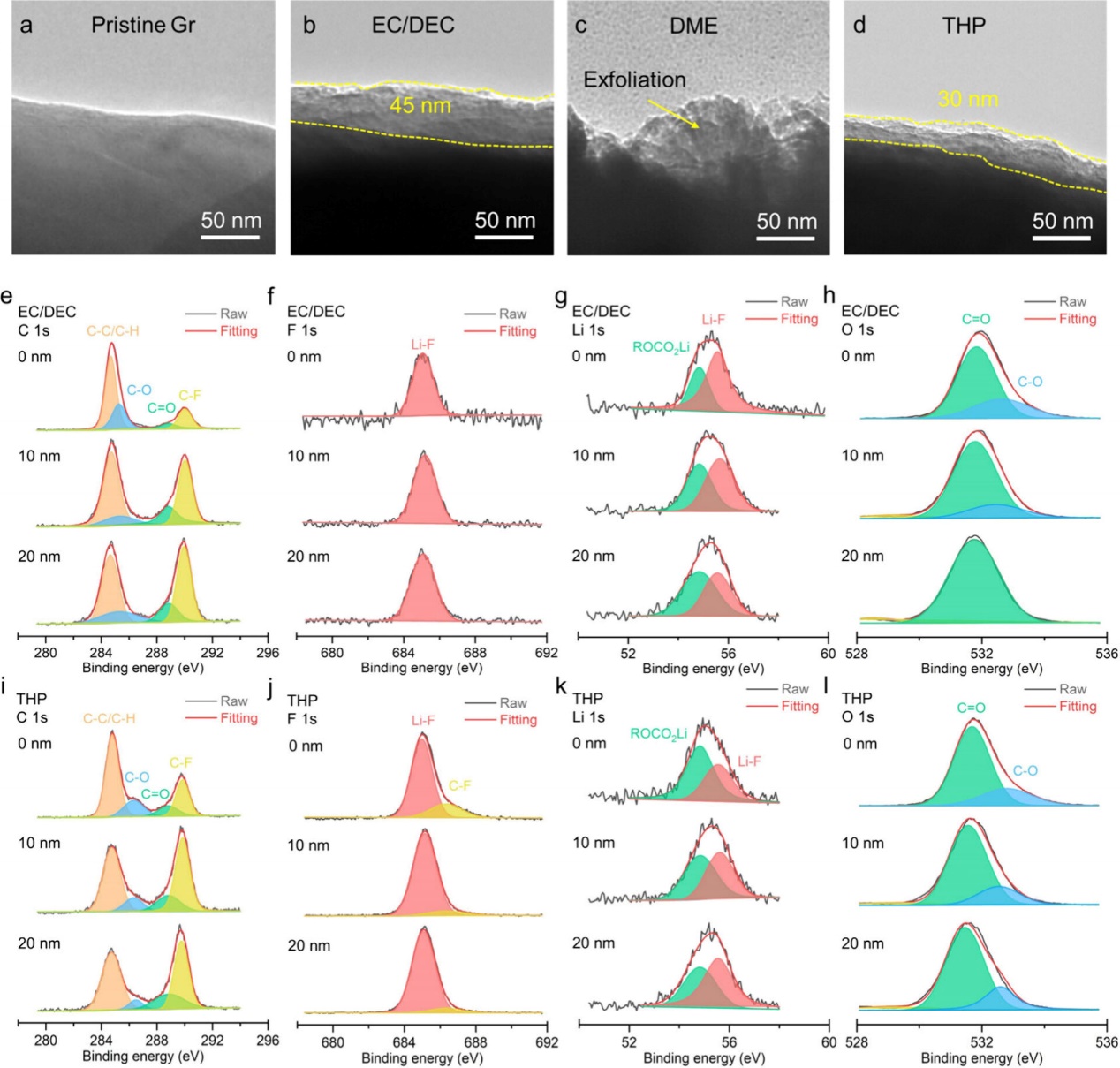
图4. (a) 原始和在 (b)EC/DEC、(c) DME 和 (d) THP 中循环的 Gr 粒子的 TEM 图像。EC/DEC 电解液中 SEI 的化学成分分析:(e) C 1s、(f) F 1s、(g) Li 1s 和 (h) O 1s 图谱。THP 电解液中 SEI 的化学成分分析:(i) C 1s、(j) F 1s、(k) Li 1s 和 (l) O 1s 图谱。
采用透射电镜技术进一步分析了循环Gr阳极的结构。新鲜的Gr粒子表面光滑,如图4a所示。在EC/DEC和THP电解质中循环后,紧凑的SEI覆盖Gr颗粒(图4b,d)。EC/ DEC和THP电解质中的SEI厚度分别为45 nm和30 nm。然而,在DME电解质中循环后,Gr显示出明显的剥落(图4c),表明没有完整的SEI形成。然后,分析了EC/ DEC和THP电解质上SEI的化学成分。检测所有电解质的有机(如ROCO2Li)和无机(如LiF)相。在C 1s光谱中,284.7、285.3、288.9和290.1eV处的峰分别属于C−C/C−H、C−O、C−O和C−F键(图4e,i)。在F 1s光谱中,Li−F和C−F键分别位于684.9和686.3 eV(图4f,j)。Li 1s光谱中54.8和55.5 eV处的峰分别代表ROCO2Li和Li−F(图4g,k)。在O 1s光谱中,531.8和532.6 eV处的峰分别对应于C=O和C−O基团(图4h, 1)。Ar+溅射后,有机相占比减少,无机相占比增加,呈双层SEI结构(有机相在上,无机相在下)。在EC/DEC和THP电解液中循环的Gr电极在C 1s和O 1s光谱中表现出相似的C−C/C−H、C−O、C−O和C−F组分。然而,THP电解质中F元素的比例(~10%)高于EC/DEC电解质(~3%),这是由于LiFSI的减少。从0、10和20 nm刻蚀深度处的元素分布来看,EC/DEC电解质中SEI的化学成分以有机相为主。相比之下,THP电解液中Gr阳极的SEI含有更多的无机物质。F 1s光谱中LiF峰面积的增加证实了SEI是富LiF的。富LiF的SEI可以抑制LixC6与电解质之间的寄生反应,有利于提高循环性能。
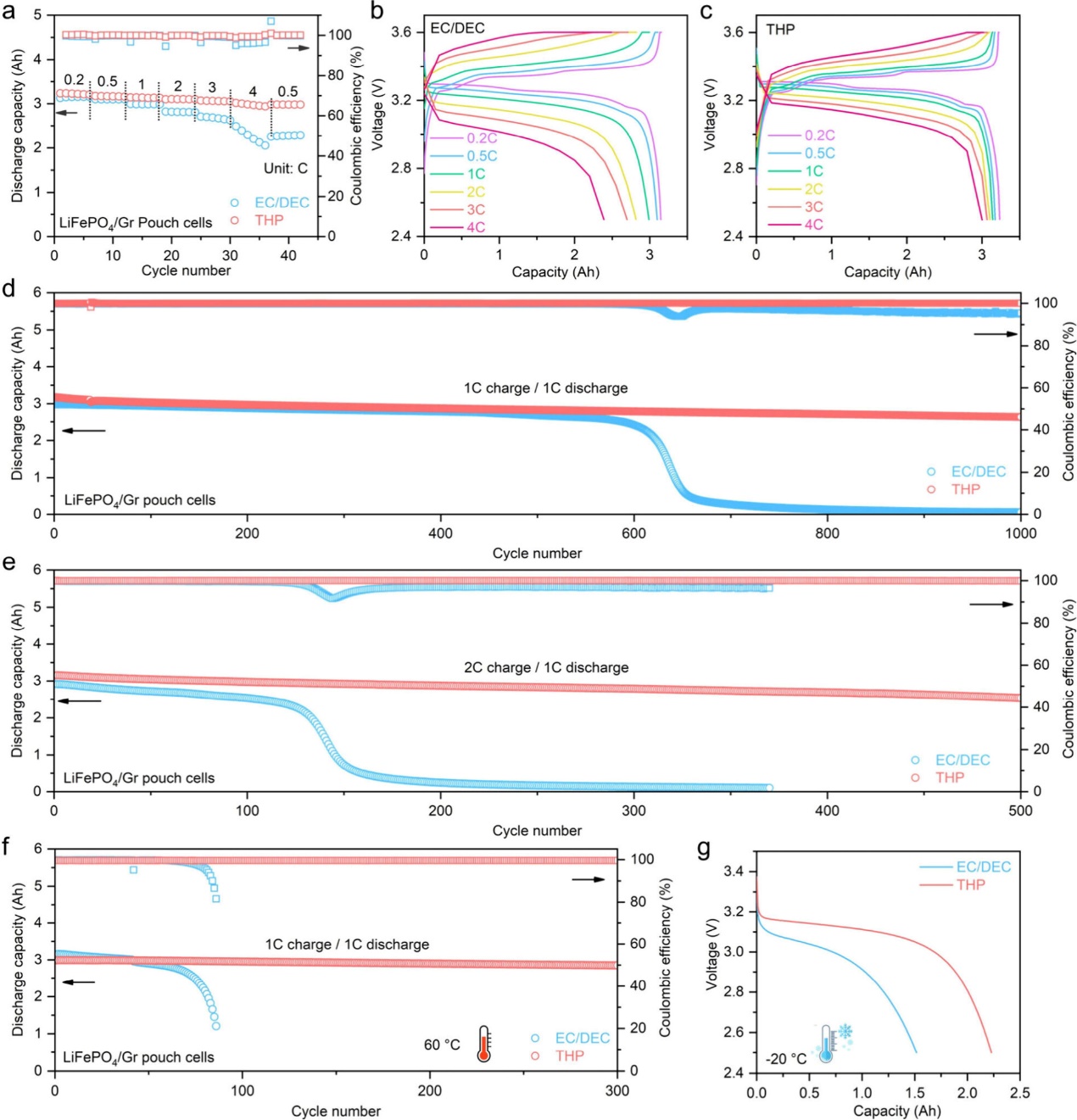
图5. LFP/Gr 软包电池的电化学性能。(a) 使用 (b) EC/DEC 和 (c) THP 电解质时的速率性能比较和电池放电/充电曲线。电池在 (d) 1 C 充电、(e) 2 C 充电和 (f) 60 ℃ 高温下的循环稳定性测试。(g) 电池在 -20 ℃ 下的放电容量。
图5a−c显示了袋状电池的容量和充放电曲线。THP软包电池在0.1、0.2、0.5、1、2和4 C充电时的放电容量分别为3.24、3.17、3.14、3.10、3.06和2.96 Ah。当电流密度恢复到0.5 C时,放电容量保持在2.98 Ah。与此形成鲜明对比的是,使用EC/DEC电解质的电池在0.1、0.2、0.5、1、2和4 C充电时的放电容量分别为3.13、3.10、2.99、2.82、2.70和2.29 Ah。当电流密度恢复到0.5 C时,放电容量仅恢复到2.28 Ah。在0.5 C的低速率下,THP软包电池显示出与使用EC/DEC电解质的电池相似的循环稳定性。然而,在1 C充电时,在THP电解质中,在1000次循环后获得了83.1%的高容量保持率(图5d)。相比之下,使用EC/DEC电解质的电池在循环600次后出现明显的容量衰减。当充电速率增加到2 C时,THP电池也可以稳定地循环500次。相比之下,使用EC/DEC电解质的电池只能在100次循环中正常工作(图5e)。良好的倍率性能归因于快速的脱溶过程。
THP软包电池也可以很好地在高温和低温下工作。在60 °C时,使用THP电解质的软包电池比EC/DEC电解质显示出更高的容量保留率(300次循环后为95.1%,80次循环后为68.2%),这归因于更少的寄生反应(图5f)。此外,THP的凝固点为- 49.2 ℃,比EC的38 ℃更低,这也有利于提高低温能力。因此,在−20 ℃的低温下,THP电解质的放电容量高达2.25 Ah,而EC /DEC电解质的电池只能显示1.52 Ah(图5g)。THP电解质由于快速的Li+脱溶过程和抑制副反应,可以改善恶劣条件下的电化学性能。
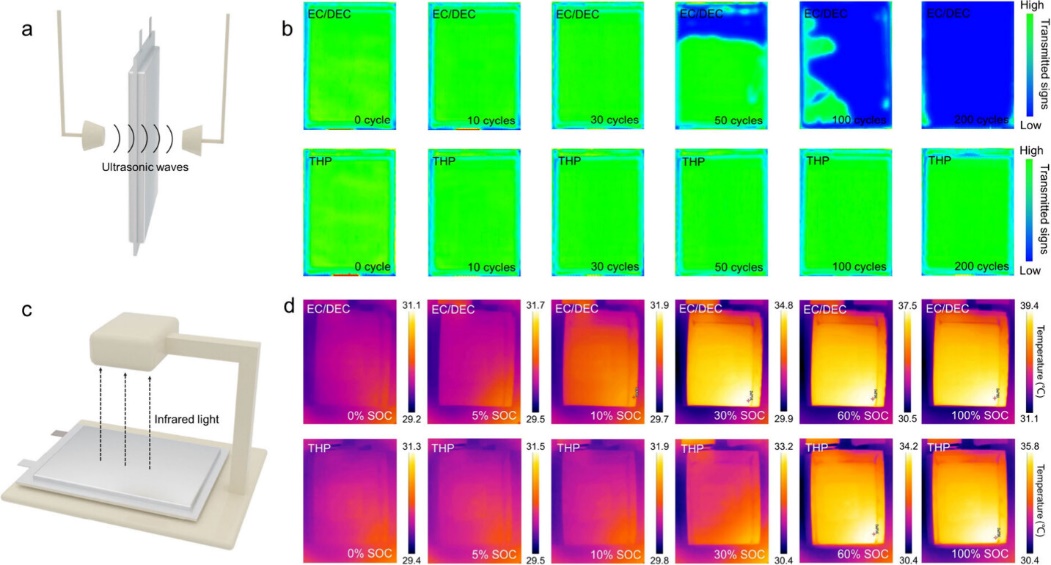
图6. (a) 超声波成像示意图。(c) 红外成像示意图。(d) 2 C 充电过程中软包电池的红外图像。
软包电池的超声分析结果如图6所示。EC/DEC细胞的图像变为蓝色,表明产生了过多的气体。同时,软包电池的厚度从4.93 mm增加到8.10 mm。与此形成鲜明对比的是,在200次循环中,THP电解质中的超声波图像仍然是绿色的,这表明电池内没有气体。电池的厚度为5.29 mm。我们使用红外成像来监测充电过程中的温度升高(图6c)。在2 C充电时,EC/DEC软包电池的表面温度高达39.4 ℃(图6d)。然而,有THP的电池的最高表面温度仅为35.4 ℃。以上结果证明,THP电解质能显著抑制软包内的寄生反应。
【研究结论】
综上所述,本研究提出六元THP作为弱溶剂溶剂在Gr负极内实现可逆和快速Li+插层。与其他环醚相比,THP的键变形程度更小,无需开环聚合即可保持结构稳定性。原位XRD和电化学分析测试证实,THP电解质能够形成可逆的Li+插层,无需溶剂共插层。从而加速了Li+的脱溶过程。使用THP基电解质的Ah级LFP/Gr软包电池具有高倍率性能(在4 C充电时为2.96 Ah),长周期稳定性(1000次循环后容量保持率为83.1%)和宽工作温度范围(- 20至60 °C)。本研究的电解液设计对于开发无碳酸盐电解质,拓宽锂离子电池在不同情况下的应用具有重要意义。
【文献链接】
A Weakly Solvating Ether Electrolyte Enables Fast-Charging and Wide-Temperature Lithium-Ion Pouch Cells.

